
¿Alguna vez te has preguntado por qué tu médico recetó un medicamento con un nombre diferente al que viste en la televisión, pero que cuesta una fracción del precio original? La respuesta no es magia ni suerte; es el resultado de un sistema regulatorio estricto diseñado para garantizar que los medicamentos genéricos sean tan seguros y efectivos como sus versiones de marca. En Estados Unidos, el Food and Drug Administration (FDA) exige estándares rigurosos antes de permitir que cualquier genérico llegue a las farmacias. Este proceso asegura que, aunque el envase sea distinto, el tratamiento que recibes funcione exactamente igual en tu cuerpo.
Entender cómo se aprueban estos medicamentos es clave para tener confianza en tu tratamiento. No se trata solo de copiar una fórmula; implica demostrar científicamente que el producto cumple con criterios precisos de identidad, fuerza, calidad, pureza y estabilidad. A continuación, desglosaremos los requisitos técnicos y legales que garantizan esta equivalencia terapéutica.
El marco legal: Ley Hatch-Waxman y la vía abreviada
La base de todo este sistema se estableció el 24 de septiembre de 1984, cuando el presidente Ronald Reagan firmó la Ley de Competencia de Precios de Medicamentos y Restauración del Término de Patentes, conocida comúnmente como Ley Hatch-Waxman. Esta legislación creó un camino de aprobación abreviado llamado Solicitud Abreviada de Nuevo Medicamento (ANDA). A diferencia de los medicamentos de marca, que deben realizar ensayos clínicos completos y costosos para probar su seguridad y eficacia desde cero, los genéricos pueden referenciarse a un medicamento de marca ya aprobado.
El objetivo principal era aumentar la competencia en el mercado para reducir los costos de las recetas sin comprometer la salud pública. Hoy en día, el Centro de Evaluación e Investigación de Medicamentos (CDER) de la FDA, específicamente su Oficina de Medicamentos Genéricos (OGD), supervisa este proceso. Según datos recientes, los medicamentos genéricos representan el 90% de todas las recetas dispensadas en EE. UU., pero solo el 23% del gasto total en medicamentos. Esto demuestra el éxito del modelo: más acceso a tratamientos equivalentes a precios accesibles.
Bioequivalencia: El corazón de la aprobación
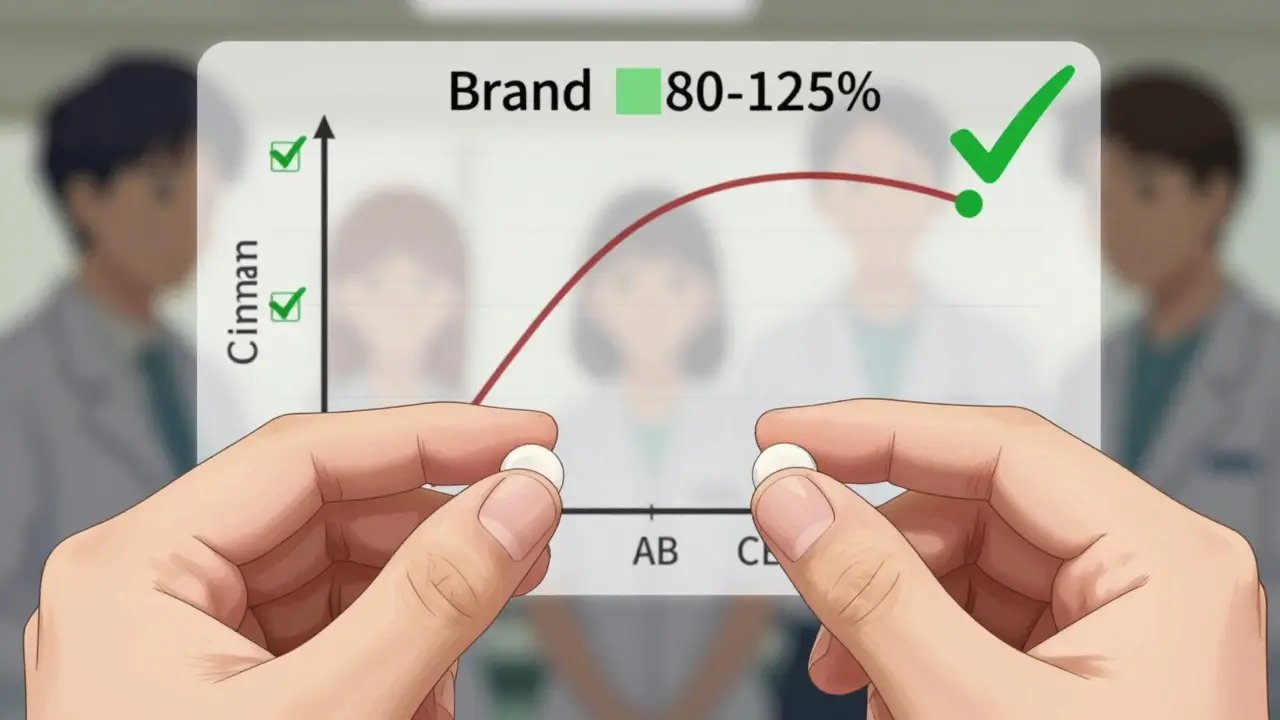
La piedra angular de la aprobación de genéricos es la bioequivalencia. Este término técnico significa que el medicamento genérico se absorbe en el cuerpo a una velocidad y en un grado similares al medicamento de referencia. Para que un genérico sea aprobado, la tasa de absorción debe caer dentro del rango del 80% al 125% de la tasa del medicamento de marca. Este margen estadístico asegura que no haya diferencias clínicamente significativas en cómo el fármaco afecta al paciente.
Los estudios de bioequivalencia suelen realizarse en voluntarios sanos. Por ejemplo, para formas orales sólidas de liberación inmediata, la FDA requiere típicamente un estudio cruzado de dos períodos con entre 24 y 36 participantes. Se miden parámetros farmacocinéticos clave como:
- Cmax: La concentración máxima del fármaco en sangre.
- AUC0-t: El área bajo la curva de concentración-tiempo hasta la última concentración medible.
- AUC0-∞: El área bajo la curva extrapolada hasta el infinito.
Todos estos valores deben caer dentro del rango aceptable del 80%-125%. Sin embargo, los productos complejos, como los de liberación modificada o inhaladores, requieren protocolos más detallados. Por ejemplo, un genérico de Ritalin LA debe demostrar patrones de liberación idénticos en intervalos específicos (0-3 horas, 3-7 horas y 7-12 horas) para asegurar que el efecto dure lo mismo que la versión original.
Calidad, Pureza y Buenas Prácticas de Manufactura
No basta con que el ingrediente activo sea correcto; el proceso de fabricación también debe ser impecable. Los fabricantes de genéricos deben cumplir con las Buenas Prácticas de Manufactura Vigentes (cGMP), definidas en los reglamentos 21 CFR Parts 210 y 211. Estas normas cubren cada aspecto de la producción, desde la limpieza de las instalaciones hasta el control de calidad de cada lote.
La FDA realiza aproximadamente 1,200 inspecciones pre-aprobación (PAI) anualmente. Si se encuentran deficiencias significativas, la aprobación se retrasa hasta que se verifiquen las acciones correctivas. Además, el sistema de Revisión Basada en Preguntas (QbR) obliga a los fabricantes a identificar y controlar los Atributos Críticos de Calidad (CQA). Esto significa que deben establecer especificaciones exhaustivas para la identidad, fuerza, calidad y pureza del producto final.
| Característica | Medicamento de Marca (NDA) | Medicamento Genérico (ANDA) |
|---|---|---|
| Datos Clínicos | Ensayos clínicos completos obligatorios | No requeridos (se basa en bioequivalencia) |
| Costo de Desarrollo | $2.6 mil millones (promedio) | $1.3 millones (promedio) |
| Tiempo de Aprobación | Varios años | 18-24 meses promedio |
| Ingredientes Activos | Nuevo principio activo | Igual al medicamento de referencia |
| Exclusividad de Mercado | Sí (protección de patentes) | No (competencia abierta tras patente) |
Desafíos con Genéricos Complejos y Fármacos de Índice Terapéutico Estrecho
No todos los genéricos son iguales en términos de dificultad de aprobación. Los llamados "genéricos complejos", como cremas tópicas, inhaladores o autoinyectores (como el EpiPen), enfrentan tasas de rechazo más altas debido a la complejidad de sus dispositivos o formulaciones. Entre 2015 y 2020, la FDA aprobó solo 3 de 27 solicitudes para un genérico del EpiPen debido a los desafíos técnicos del dispositivo.
Otro punto crítico son los fármacos de índice terapéutico estrecho. Estos son medicamentos donde pequeñas variaciones en la dosis pueden causar efectos adversos graves o falta de eficacia, como la warfarina o la levothyroxina. Algunos expertos han criticado el rango estándar del 80%-125% para estos casos. En respuesta, la FDA ha implementado límites más estrictos. Por ejemplo, para la levothyroxina sódica, se exige un rango de bioequivalencia del 95%-105%, asegurando una consistencia mucho mayor para pacientes con condiciones delicadas.
El Proceso Práctico: Costos, Tiempos y Recursos
Llevar un genérico al mercado requiere experiencia regulatoria significativa. La guía paso a paso de la FDA indica que las actividades previas a la presentación, como el desarrollo del producto y la planificación de estudios de bioequivalencia, toman entre 18 y 24 meses. Una solicitud ANDA típica contiene entre 5,000 y 10,000 páginas de documentación, incluyendo datos de química, manufactura y controles (CMC).
Según el programa de Tasas de Usuarios de Medicamentos Genéricos (GDUFA), la mayoría de las aplicaciones estándar reciben una revisión en el primer ciclo dentro de 10 meses. Sin embargo, menos del 10% de las solicitudes de genéricos se aprueban en el primer ciclo de revisión, comparado con el 90% de los nuevos medicamentos de marca. Esta discrepancia se debe a la complejidad técnica y a inconsistencias en la manufactura. Los factores de éxito incluyen la participación temprana con la FDA a través del programa Pre-ANDA y la inversión en robustos sistemas de gestión de calidad.
Perspectiva de Mercado y Futuro Regulatorio
El impacto económico de los genéricos es enorme. En 2022, representaron $373 mil millones en ahoros anuales en el sistema de salud estadounidense. Empresas líderes como Teva, Viatris y Sandoz dominan gran parte del mercado, pero la competencia sigue siendo intensa. El futuro apunta hacia la aprobación de más genéricos complejos. La FDA tiene como objetivo aprobar el 50% de las aplicaciones de genéricos complejos dentro de dos ciclos de revisión para 2027, un aumento significativo respecto al actual 28%.
A pesar de los avances, persisten desafíos como las estrategias de "evergreening" (perpetuación de patentes) por parte de las compañías de marca, que pueden retrasar la entrada de genéricos al mercado incluso después de obtener la aprobación regulatoria. Estudios de la Comisión Federal de Comercio indican un retraso promedio de 2.4 años entre la aprobación de la ANDA y la entrada real al mercado debido a litigios de patentes.
¿Son los medicamentos genéricos realmente tan efectivos como los de marca?
Sí, la evidencia científica respalda su equivalencia. La AMA concluyó en 2021 que los estándares de bioequivalencia son científicamente sólidos, mostrando resultados clínicos equivalentes en el 98.7% de las categorías terapéuticas durante 15 años de vigilancia post-comercialización. La FDA exige que contengan el mismo ingrediente activo, dosis y forma de administración.
¿Por qué algunos médicos prefieren recetar medicamentos de marca?
A veces hay preferencia por marcas en casos muy específicos, como fármacos de índice terapéutico estrecho donde la consistencia extrema es vital, o cuando existen problemas de suministro de genéricos. También puede haber influencia de marketing o hábitos establecidos. Sin embargo, para la gran mayoría de los tratamientos, los genéricos son la opción recomendada por su costo-efectividad y seguridad comprobada.
¿Qué es la Ley Hatch-Waxman y por qué es importante?
Firmada en 1984, esta ley creó el proceso abreviado (ANDA) para aprobar genéricos sin necesidad de repetir ensayos clínicos costosos, basándose en la equivalencia con un medicamento ya aprobado. Fue crucial para democratizar el acceso a medicamentos al fomentar la competencia y reducir drásticamente los precios.
¿Cuánto tiempo tarda en aprobarse un medicamento genérico?
El tiempo promedio desde la presentación hasta la aprobación es de aproximadamente 32.7 meses para genéricos convencionales, aunque puede extenderse a 47.2 meses para genéricos complejos. La fase previa de desarrollo y preparación suele tomar entre 18 y 24 meses adicionales.
¿Qué diferencia a un genérico complejo de uno convencional?
Los genéricos complejos incluyen productos como inhaladores, parches transdérmicos, inyectables de liberación prolongada o cremas tópicas. Su desafío radica en demostrar que el mecanismo de entrega del fármaco al cuerpo es idéntico al de la marca, lo cual requiere estudios de bioequivalencia más sofisticados y costosos que las pastillas orales simples.
¿Puedo cambiar entre diferentes marcas de genéricos libremente?
Generalmente sí, ya que todos cumplen con los mismos estándares de bioequivalencia de la FDA. Sin embargo, si notas algún cambio en cómo te sientes al cambiar de fabricante, consulta a tu médico. Aunque es raro, algunas personas pueden ser sensibles a excipientes (ingredientes inactivos) diferentes entre marcas.





Categorías