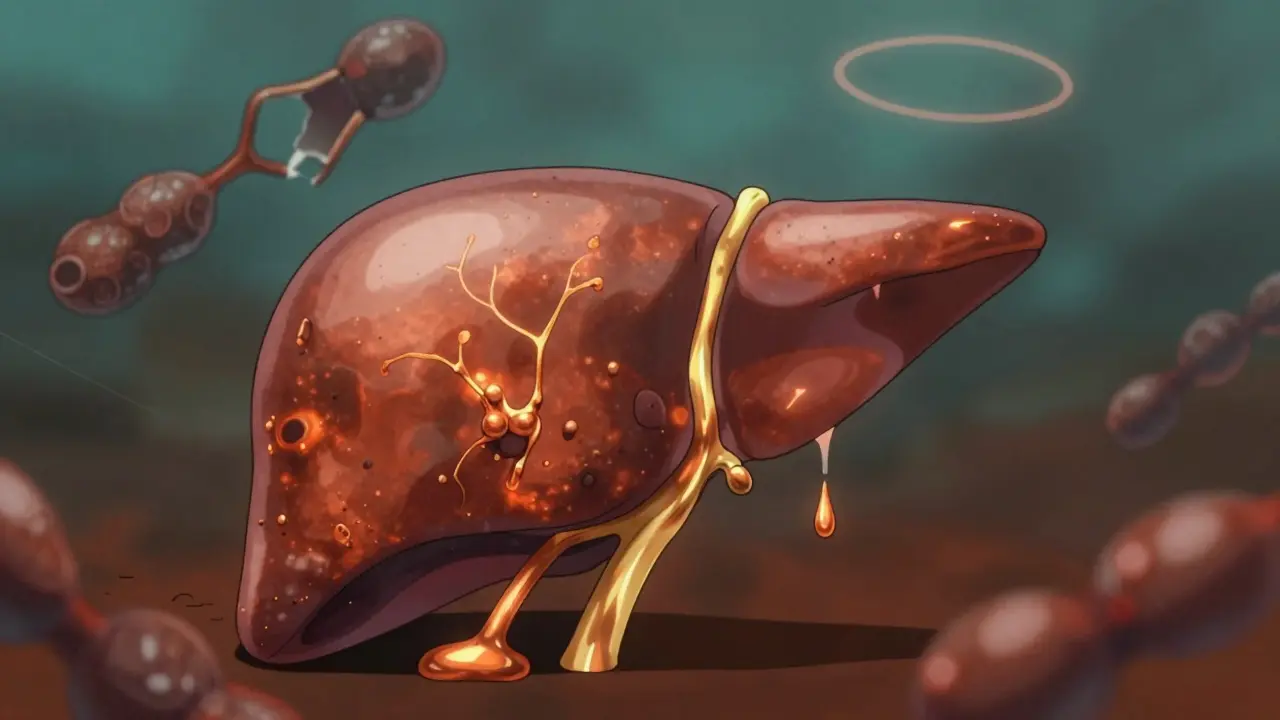
La enfermedad de Wilson es una afección rara pero grave que hace que el cuerpo acumule cobre tóxico. A primera vista, puede parecer un problema hepático común: fatiga, ictericia, dolor abdominal. Pero detrás de esos síntomas hay una falla molecular silenciosa: el cuerpo ya no puede eliminar el cobre. Si no se trata, este metal se acumula en el hígado, el cerebro y los ojos, causando daños irreversibles. Lo que hace única a esta enfermedad es que, a diferencia de otras enfermedades del hígado, la enfermedad de Wilson se puede controlar con medicamentos si se detecta a tiempo.
¿Qué pasa cuando el cuerpo no elimina el cobre?
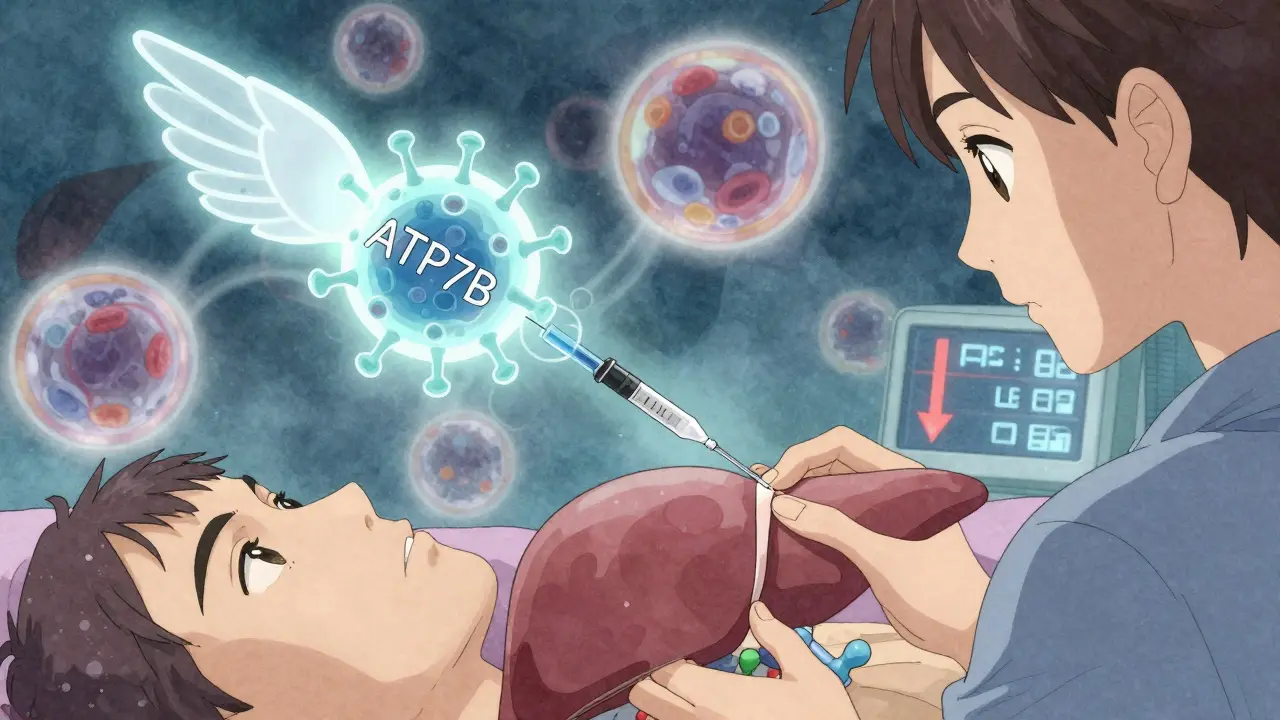
Tu cuerpo necesita cobre. Lo usas para formar glóbulos rojos, mantener tus nervios sanos y producir energía en las células. Normalmente, lo absorbes por la comida, lo transportas al hígado y luego lo expulsas por la bilis. Todo esto lo hace una proteína llamada ATP7B. Pero en la enfermedad de Wilson, hay una mutación en el gen que produce esa proteína. No funciona. Y el cobre se queda.
Al principio, el hígado intenta protegerse. Almacena el exceso de cobre en una molécula llamada metalotioneína. Pero esa capacidad tiene límites. Cuando se satura, el cobre empieza a filtrarse a la sangre como cobre libre -no unido a la ceruloplasmina-. Ese cobre libre es el verdadero enemigo. Viaja por el cuerpo y se acumula donde no debería: en el cerebro, especialmente en la región llamada ganglios basales, y en los ojos, donde forma anillos marrones alrededor de la córnea, conocidos como anillos de Kayser-Fleischer. Estos anillos son una señal clave: están presentes en el 95% de las personas con síntomas neurológicos.
¿Cómo se diagnostica una enfermedad que se parece a otras?
Una de las mayores dificultades es que la enfermedad de Wilson se confunde fácilmente con hepatitis autoinmune o enfermedad hepática grasa. Muchos pacientes esperan años antes de recibir un diagnóstico correcto. En promedio, la demora es de 2,7 años. Por eso, los médicos usan una combinación de pruebas.
- Ceruloplasmina en sangre: Normalmente entre 20 y 50 mg/dL. En la enfermedad de Wilson, suele estar por debajo de 20 mg/dL.
- Cobre en orina de 24 horas: Más de 100 μg/día es un indicador fuerte. En casos neurológicos, puede ser más bajo, pero aún fuera de rango.
- Cobre libre en sangre: Si supera los 10 μg/dL, es una señal de toxicidad.
- Anillos de Kayser-Fleischer: Se ven con una lámpara especial llamada lámpara de hendidura. Su presencia es casi diagnóstica si hay síntomas.
- Análisis genético: Detectar mutaciones en el gen ATP7B confirma el diagnóstico, especialmente en niños o cuando otros resultados son ambiguos.
El sistema de puntuación actual, actualizado en 2023, permite clasificar la probabilidad de enfermedad. Una mutación confirmada en ATP7B suma 4 puntos. Si sumas 4 o más, el diagnóstico es definitivo.
Terapia de quelación: ¿Cómo se saca el cobre del cuerpo?
La clave para sobrevivir con enfermedad de Wilson es eliminar el cobre sin causar más daño. Eso se hace con quelantes: medicamentos que se unen al cobre y lo llevan fuera del cuerpo por la orina.
El más antiguo y usado es la D-penicilamina. Funciona bien, pero tiene un problema grave: en 20-50% de los casos, empeora los síntomas neurológicos al principio del tratamiento. Por eso, muchos médicos la combinan con zinc durante las primeras semanas. El zinc bloquea la absorción de cobre en el intestino, lo que ayuda a calmar la tormenta.
Una alternativa es la trientina. Es menos tóxica para el sistema nervioso, pero cuesta casi seis veces más: unos 1.850 dólares al mes en EE.UU., frente a los 300 de la penicilamina. No es tan accesible, pero es la opción preferida cuando la penicilamina causa reacciones graves.
El zinc acetato no es un quelante, pero juega un papel vital. Actúa como escudo: hace que el intestino produzca metalotioneína, que atrapa el cobre de la comida antes de que entre al torrente sanguíneo. Se usa principalmente para mantenimiento después de que el cobre se ha reducido. Es eficaz en el 92% de los casos si se mantiene el cobre libre por debajo de 10 μg/dL.

Los desafíos reales de vivir con tratamiento
Tomar medicamentos para la enfermedad de Wilson no es como tomar una pastilla para el dolor de cabeza. Es un régimen complejo. La penicilamina y la trientina deben tomarse con el estómago vacío, 1 hora antes o 2 horas después de comer. Muchos pacientes se olvidan, se sienten mareados o tienen un sabor metálico constante en la boca. El 38% reporta ese sabor desagradable. El 42% tiene náuseas.
Además, hay que seguir una dieta baja en cobre. Menos de 1 mg al día. Eso significa evitar hígado, mariscos, nueces, chocolate, champiñones y agua de pozos profundos. Muchos pacientes se sienten aislados en restaurantes o reuniones familiares. Una encuesta de la Fundación de la Enfermedad de Wilson mostró que el 89% de los pacientes tiene dificultades para mantener esta dieta sin quedar deficiente en otros nutrientes.
Y no todo el mundo responde igual. Algunos pacientes, como uno que compartió en Reddit, mejoraron tras cambiar de penicilamina a zinc. Otros desarrollan lupus por efecto secundario de la penicilamina. El 22% de los usuarios de este medicamento lo experimentan. La trientina, por su parte, puede causar deficiencia de hierro en el 35% de los casos.
Nuevos tratamientos y esperanza en el horizonte
La investigación no se detiene. En 2023, un nuevo fármaco llamado CLN-1357, un polímero que atrapa el cobre, redujo el cobre libre en la sangre en un 82% en solo 12 semanas, sin empeorar los síntomas neurológicos. Eso es un avance enorme.
El medicamento WTX101, aprobado con designación de terapia de ruptura por la FDA en enero de 2023, mostró una eficacia del 91% en prevenir el deterioro neurológico, superando a la trientina. Y en Europa, ya se usa Decuprate®, una forma mejorada de tetratiomolibdato, que cruza mejor la barrera hematoencefálica.
Lo más prometedor es la terapia génica. En 2023, un pequeño estudio con seis pacientes recibió una versión sana del gen ATP7B mediante un virus modificado. Los resultados iniciales fueron seguros. Aunque todavía está en fase temprana, es la primera vez que se intenta corregir la causa raíz, no solo sus síntomas.
¿Qué pasa si no se trata?
La enfermedad de Wilson es fatal si no se trata. El cobre destruye el hígado hasta causar cirrosis o insuficiencia hepática. En el cerebro, provoca temblores, rigidez, dificultad para hablar y tragar, y en casos avanzados, demencia o coma. Pero si se detecta antes de que se produzcan daños graves, la expectativa de vida es normal. La clave es la detección temprana.
En países con recursos limitados, la demora en el diagnóstico puede superar los cinco años. Allí, muchos pacientes mueren sin saber qué les pasaba. Pero en lugares donde se hacen las pruebas adecuadas, la historia es diferente. Con tratamiento, una persona con enfermedad de Wilson puede trabajar, tener hijos, viajar, vivir una vida plena.
¿Qué debes hacer si sospechas que podrías tenerla?
Si tienes síntomas hepáticos o neurológicos inexplicables -especialmente si tienes menos de 35 años- y alguien en tu familia tiene enfermedad de Wilson, pide una evaluación. No esperes a que empeore. Pide:
- Una prueba de ceruloplasmina en sangre
- Una medición de cobre en orina de 24 horas
- Una revisión ocular con lámpara de hendidura
- Un análisis genético si hay dudas
No es una enfermedad común, pero es tratable. Y en medicina, eso es lo más valioso que puede haber.
¿La enfermedad de Wilson se hereda?
Sí. Es una enfermedad autosómica recesiva. Esto significa que una persona debe heredar dos copias defectuosas del gen ATP7B, una de cada padre. Si solo heredas una, eres portador pero no tendrás la enfermedad. Dos portadores tienen un 25% de probabilidad de tener un hijo con la enfermedad.
¿Puedo tomar suplementos de cobre si tengo enfermedad de Wilson?
No. Cualquier suplemento de cobre está contraindicado. Incluso multivitamínicos que contienen cobre deben evitarse. Tu cuerpo ya tiene demasiado. Lo que necesitas es eliminarlo, no añadir más.
¿Cuánto tiempo debo tomar los medicamentos?
Para siempre. La enfermedad de Wilson no se cura, pero se controla. Dejar de tomar los medicamentos, aunque te sientas bien, hace que el cobre vuelva a acumularse. El tratamiento es de por vida, pero con adherencia, puedes vivir sin síntomas.
¿Los anillos de Kayser-Fleischer desaparecen con el tratamiento?
Sí, pero lentamente. Con tratamiento adecuado, los anillos pueden desvanecerse en meses o años. Sin embargo, no siempre desaparecen por completo. Su presencia o ausencia no indica si el tratamiento funciona; lo que importa son los niveles de cobre en sangre y orina.
¿Puedo tener hijos si tengo enfermedad de Wilson?
Sí. Las mujeres con enfermedad de Wilson bien controlada pueden embarazarse y tener hijos sanos. Es importante mantener los niveles de cobre estables durante el embarazo. El tratamiento debe ajustarse bajo supervisión médica, pero no hay contraindicaciones absolutas.




