Evaluar la bioequivalencia de fármacos altamente variables es uno de los mayores desafíos en el desarrollo de genéricos. Si un medicamento tiene una variabilidad intra-sujeto (ISCV) superior al 30%, los diseños tradicionales de cruzamiento 2x2 fallan. Requieren cientos de voluntarios, son costosos y a menudo inviables. Aquí es donde entran los diseños de estudio replicados: la única forma práctica y científicamente sólida de demostrar que un genérico es equivalente a su marca de referencia cuando el fármaco es inestable en el cuerpo.
¿Por qué los diseños tradicionales no funcionan con fármacos altamente variables?
Imagina que pruebas un nuevo genérico de un fármaco como warfarina o levo-tiroxina. El cuerpo lo absorbe de forma muy distinta cada vez: en un día la concentración en sangre puede ser el doble que en otro, incluso con la misma dosis. Esto no es error de medición. Es variabilidad biológica real. En un estudio 2x2 estándar (un grupo toma el genérico, luego el original; otro lo hace al revés), esa variabilidad se confunde con diferencias entre fármacos. El resultado: falsos negativos. Tu genérico puede ser perfecto, pero el estudio lo rechaza porque el ruido supera la señal.
Esto no es teoría. En 2018, el 41% de las solicitudes de bioequivalencia para fármacos altamente variables fueron rechazadas por la FDA porque usaban diseños no replicados. La mayoría de esos rechazos se debían a que los estudios no tenían potencia estadística suficiente. La solución no es aumentar el tamaño de la muestra sin límites. Es cambiar el diseño.
¿Qué son los diseños replicados y cómo funcionan?
Un diseño replicado significa que los participantes reciben más de una dosis del mismo fármaco en diferentes periodos. Esto permite separar la variabilidad del fármaco de la variabilidad entre personas. Hay tres tipos principales:
- Diseño de replicación completa (4 periodos): TRRT o RTRT. Cada sujeto toma el fármaco de prueba dos veces y el de referencia dos veces. Permite estimar la variabilidad tanto del genérico (CVwT) como del original (CVwR).
- Diseño de replicación completa (3 periodos): TRT o RTR. Cada sujeto toma el genérico una vez y el original dos veces (o viceversa). Solo estima la variabilidad del fármaco de referencia (CVwR).
- Diseño de replicación parcial (3 periodos): TRR, RTR, RRT. Similar al anterior, pero con secuencias mixtas. También solo estima CVwR.
La clave está en el cálculo. En lugar de usar límites fijos de bioequivalencia (80-125%), los diseños replicados permiten escalar esos límites según la variabilidad real del fármaco de referencia. Esto se llama reference-scaled average bioequivalence (RSABE). Si el CVwR es del 30%, los límites se amplían hasta 100-112%. Si es del 50%, pueden llegar hasta 69.8-143.2%. Esto no es un truco. Es lógica estadística: si el fármaco original varía mucho, es razonable aceptar una mayor variación en el genérico, siempre que su comportamiento promedio sea similar.
¿Cuándo usar cada diseño? Guía práctica
No todos los diseños replicados son iguales. La elección depende de la variabilidad esperada y del tipo de fármaco.
- ISCV < 30%: Usa el diseño 2x2 estándar. Es más simple, barato y suficiente.
- ISCV entre 30% y 50%: El diseño TRT/RTR de 3 periodos es el más usado. Ofrece un buen equilibrio entre potencia estadística y viabilidad operativa. Según una encuesta de 47 CROs en 2023, el 83% lo considera óptimo.
- ISCV > 50% o fármacos de índice terapéutico estrecho (NTI): Usa el diseño TRRT/RTRT de 4 periodos. La FDA lo exige explícitamente para medicamentos como warfarina, fenitoína o levo-tiroxina. Aquí, estimar la variabilidad del genérico es crítico para garantizar seguridad.
La FDA requiere al menos 12 sujetos en la secuencia RTR para que un diseño de 3 periodos sea válido. Eso significa un mínimo de 24 sujetos en total, distribuidos equitativamente entre secuencias. Pero no basta con el número mínimo. Debido a la tasa de abandono (15-25% en estudios de múltiples periodos), debes reclutar un 20-30% más. Si planeas 24 sujetos, recluta 30. Si planeas 36, recluta 45.
Costos, duración y desafíos operativos
Los diseños replicados no son más fáciles. Son más largos, más caros y más complejos.
Una secuencia TRT/RTR dura al menos 21 días, pero puede extenderse a 6-8 semanas si el fármaco tiene una vida media larga. Los periodos de lavado entre dosis deben ser lo suficientemente largos para eliminar completamente el fármaco del cuerpo. Si no lo son, los resultados se contaminan. Muchos estudios fracasan por esto.
El abandono de sujetos es otro problema real. Un estudio en Reddit reportó una tasa de abandono del 30% en un diseño de 4 periodos para un fármaco de vida media larga. El resultado: 8 semanas de retraso y $187,000 adicionales en costos. La solución: ofrecer incentivos claros, mantener comunicación constante y elegir fármacos con periodos de lavado manejables.
El análisis estadístico también es un obstáculo. No puedes usar Excel o software genérico. Necesitas herramientas especializadas como Phoenix WinNonlin, R con el paquete replicateBE (versión 0.12.1, descargado más de 1,200 veces en 2024), o SAS con modelos de efectos mixtos. Aprenderlo lleva entre 80 y 120 horas de formación técnica. Sin esta habilidad, incluso un buen estudio puede rechazarse por análisis incorrecto.
Comparación: Replicado vs. Estándar
La diferencia en eficiencia es abismal. Aquí tienes una comparación real:
| ISCV | Diferencia de formulación | Diseño 2x2 estándar | Diseño replicado (3 periodos) |
|---|---|---|---|
| 30% | 5% | 38 sujetos | 24 sujetos |
| 40% | 8% | 68 sujetos | 36 sujetos |
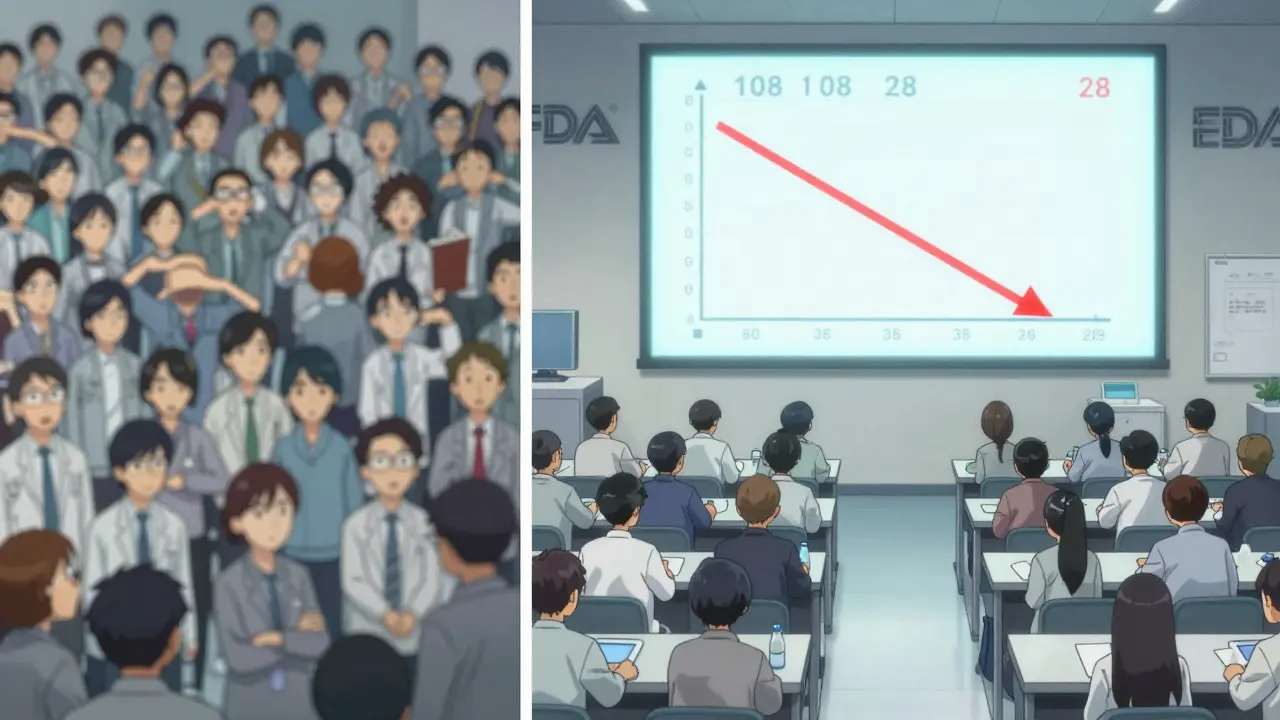
| 50% | 10% | 108 sujetos | 28 sujetos |
En el caso del 50% de ISCV, el diseño replicado reduce el tamaño de muestra en un 74%. Eso significa menos voluntarios, menos costos, menos tiempo y, lo más importante, menos riesgo de fracaso. En 2023, el 79% de los estudios replicados bien ejecutados fueron aprobados por la FDA, frente al 52% de los estudios no replicados para fármacos altamente variables.

Regulación global: FDA, EMA y tendencias futuras
La FDA y la EMA aceptan diseños replicados, pero con diferencias. La FDA prefiere diseños de 4 periodos para NTI y recomienda el TRRT/RTRT. La EMA acepta con igual rigor el diseño de 3 periodos TRT/RTR. Esto crea confusión para las empresas que buscan aprobación global. Un estudio que funciona en EE.UU. puede ser rechazado en Europa si no cumple con el formato preferido.
La ICH está trabajando en una guía conjunta (E14/S6(R1)) para armonizar estos enfoques, con resultados esperados en el tercer trimestre de 2024. Mientras tanto, las agencias están adoptando nuevas herramientas. La FDA ya acepta métodos bayesianos en ciertos casos, y empresas como Pfizer están probando inteligencia artificial para predecir el tamaño de muestra ideal usando datos históricos. En pruebas, estos modelos alcanzaron un 89% de precisión.
El mercado de estudios de bioequivalencia creció a $2.8 mil millones en 2023. El 35% de los estudios para fármacos altamente variables ya usan diseños replicados, frente al 18% en 2019. WuXi AppTec, PPD y Charles River dominan el mercado, pero las pequeñas CROs con experiencia estadística, como BioPharma Services, están ganando terreno.
Errores comunes y cómo evitarlos
Los estudios replicados no son infalibles. Tres errores frecuentes arruinan incluso los mejores protocolos:
- Períodos de lavado insuficientes: No limpiar completamente el fármaco entre dosis contamina los resultados. Usa datos de vida media y modelos farmacocinéticos para calcularlo.
- Abandono de sujetos: No reclutar suficientes voluntarios adicionales. El 20-30% extra no es un lujo, es una necesidad.
- Modelo estadístico incorrecto: Usar ANOVA en lugar de modelos de efectos mixtos. El RSABE requiere análisis específicos. Si no lo haces bien, la FDA lo rechazará.
Un gerente de operaciones clínicas compartió en el foro BEBAC: "Nuestro estudio de levo-tiroxina con 42 sujetos en diseño TRT/RTR pasó en la primera presentación. Antes, con 98 sujetos en diseño 2x2, fallamos tres veces." Esa es la diferencia entre hacerlo bien y hacerlo mal.
¿Qué debes hacer ahora?
Si estás desarrollando un genérico para un fármaco con ISCV > 30%, no pienses en el diseño 2x2. No es una opción viable. Empieza por:
- Revisar la literatura o datos previos para estimar el ISCV del fármaco de referencia.
- Seleccionar el diseño replicado adecuado: 3 periodos para ISCV 30-50%, 4 periodos para >50% o NTI.
- Contratar a un estadístico con experiencia en replicateBE o WinNonlin. No lo hagas tú mismo si no tienes formación específica.
- Reclutar un 25% más de sujetos de lo que calculas como mínimo.
- Documentar cada decisión del protocolo con justificación científica y regulatoria.
Los diseños replicados no son el futuro. Ya son el presente. Y en 2026, no hay vuelta atrás. Quien no los entienda, no podrá llevar un genérico al mercado.
¿Qué significa ISCV y por qué es tan importante en bioequivalencia?
ISCV significa coeficiente de variación intra-sujeto. Mide cuánto varía la concentración de un fármaco en la sangre de una misma persona cuando se toma la misma dosis en diferentes ocasiones. Si el ISCV es mayor del 30%, el fármaco es considerado altamente variable. En esos casos, los límites tradicionales de bioequivalencia (80-125%) no son apropiados, porque la variabilidad natural del fármaco es tan alta que cualquier diferencia pequeña entre genérico y marca se pierde en el ruido. Por eso se usa RSABE: para ajustar los límites según la variabilidad real, no según un número fijo.
¿Puedo usar un diseño replicado para un fármaco con ISCV de 25%?
Técnicamente sí, pero no es recomendable. Si el ISCV es menor del 30%, el diseño 2x2 estándar es más eficiente, económico y aceptado por todas las agencias. Usar un diseño replicado en este caso añade complejidad innecesaria, aumenta costos y puede generar dudas regulatorias. Las guías de la FDA y la EMA indican claramente que los diseños replicados están pensados para ISCV ≥ 30%. No se usan para fármacos con baja variabilidad.
¿Cuál es la diferencia entre diseño de replicación completa y parcial?
El diseño de replicación completa (como TRT o TRRT) permite estimar la variabilidad tanto del fármaco de prueba como del de referencia. El diseño de replicación parcial (como TRR o RTR) solo estima la variabilidad del fármaco de referencia. La FDA acepta ambos para RSABE, pero solo el diseño completo es obligatorio para fármacos de índice terapéutico estrecho, porque necesitas saber si el genérico también es variable. La EMA acepta el parcial en la mayoría de los casos, lo que lo hace más popular en Europa.
¿Por qué la FDA exige TRRT/RTRT para warfarina?
Porque la warfarina es un fármaco de índice terapéutico estrecho (NTI), donde una pequeña diferencia en la concentración puede causar sangrado o trombosis. Además, tiene una variabilidad muy alta (ISCV > 50%). En este caso, no basta con saber que el genérico es similar en promedio. Tienes que asegurarte de que su variabilidad no sea mayor que la del original. Solo el diseño TRRT/RTRT permite estimar la variabilidad del genérico (CVwT) y compararla directamente con la del original (CVwR). Si CVwT es mayor, el estudio falla, aunque el promedio sea igual.
¿Qué software se usa para analizar estudios replicados?
El software estándar es Phoenix WinNonlin, SAS y el paquete R replicateBE. De estos, replicateBE es el más usado en la industria por ser gratuito, transparente y validado por la FDA. Su documentación incluye ejemplos reales de cálculos de RSABE y se actualiza anualmente. Muchas CROs lo usan como estándar interno. No se recomienda usar Excel, SPSS o programas genéricos, porque no implementan correctamente los modelos de efectos mixtos necesarios para RSABE.
¿Es más difícil obtener aprobación con un diseño replicado?
No, al contrario. Los estudios replicados bien diseñados tienen una tasa de aprobación del 79%, frente al 52% de los estudios no replicados para fármacos altamente variables. El problema no es el diseño, sino la ejecución. Si el protocolo es incorrecto, el análisis es mal hecho, o hay demasiados abandonos, el estudio fracasará. Pero si se sigue la guía regulatoria, el diseño replicado es la mejor forma de asegurar aprobación, no de evitarla.




